Supervised learning is an area of machine learning where the chosen algorithm tries to fit a target using the given input. A set of training data that contains labels is supplied to the algorithm. Based on a massive set of data, the algorithm will learn a rule that it uses to predict the labels for new observations. In other words, supervised learning algorithms are provided with historical data and asked to find the relationship that has the best predictive power.
There are two varieties of supervised learning algorithms: regression and classification algorithms. Regression-based supervised learning methods try to predict outputs based on input variables. Classification-based supervised learning methods identify which category a set of data items belongs to. Classification algorithms are probability-based, meaning the outcome is the category for which the algorithm finds the highest probability that the dataset belongs to it. Regression algorithms, in contrast, estimate the outcome of problems that have an infinite number of solutions (continuous set of possible outcomes).
In the context of finance, supervised learning models represent one of the most-used class of machine learning models. Many algorithms that are widely applied in algorithmic trading rely on supervised learning models because they can be efficiently trained, they are relatively robust to noisy financial data, and they have strong links to the theory of finance.
Regression-based algorithms have been leveraged by academic and industry researchers to develop numerous asset pricing models. These models are used to predict returns over various time periods and to identify significant factors that drive asset returns. There are many other use cases of regression-based supervised learning in portfolio management and derivatives pricing.
Classification-based algorithms, on the other hand, have been leveraged across many areas within finance that require predicting a categorical response. These include fraud detection, default prediction, credit scoring, directional forecast of asset price movement, and Buy/Sell recommendations. There are many other use cases of classification-based supervised learning in portfolio management and algorithmic trading.
Many use cases of regression-based and classification-based supervised machine learning are presented in Chapters 5 and 6.
Python and its libraries provide methods and ways to implement these supervised learning models in few lines of code. Some of these libraries were covered in Chapter 2. With easy-to-use machine learning libraries like Scikit-learn and Keras, it is straightforward to fit different machine learning models on a given predictive modeling dataset.
In this chapter, we present a high-level overview of supervised learning models. For a thorough coverage of the topics, the reader is referred to Hands-On Machine Learning with Scikit-Learn, Keras, and TensorFlow, 2nd Edition, by Aurélien Géron (O’Reilly).
The following topics are covered in this chapter:
- Basic concepts of supervised learning models (both regression and classification).
- How to implement different supervised learning models in Python.
- How to tune the models and identify the optimal parameters of the models using grid search.
- Overfitting versus underfitting and bias versus variance.
- Strengths and weaknesses of several supervised learning models.
- How to use ensemble models, ANN, and deep learning models for both regression and classification.
- How to select a model on the basis of several factors, including model performance.
- Evaluation metrics for classification and regression models.
- How to perform cross validation.
Supervised Learning Models: An Overview
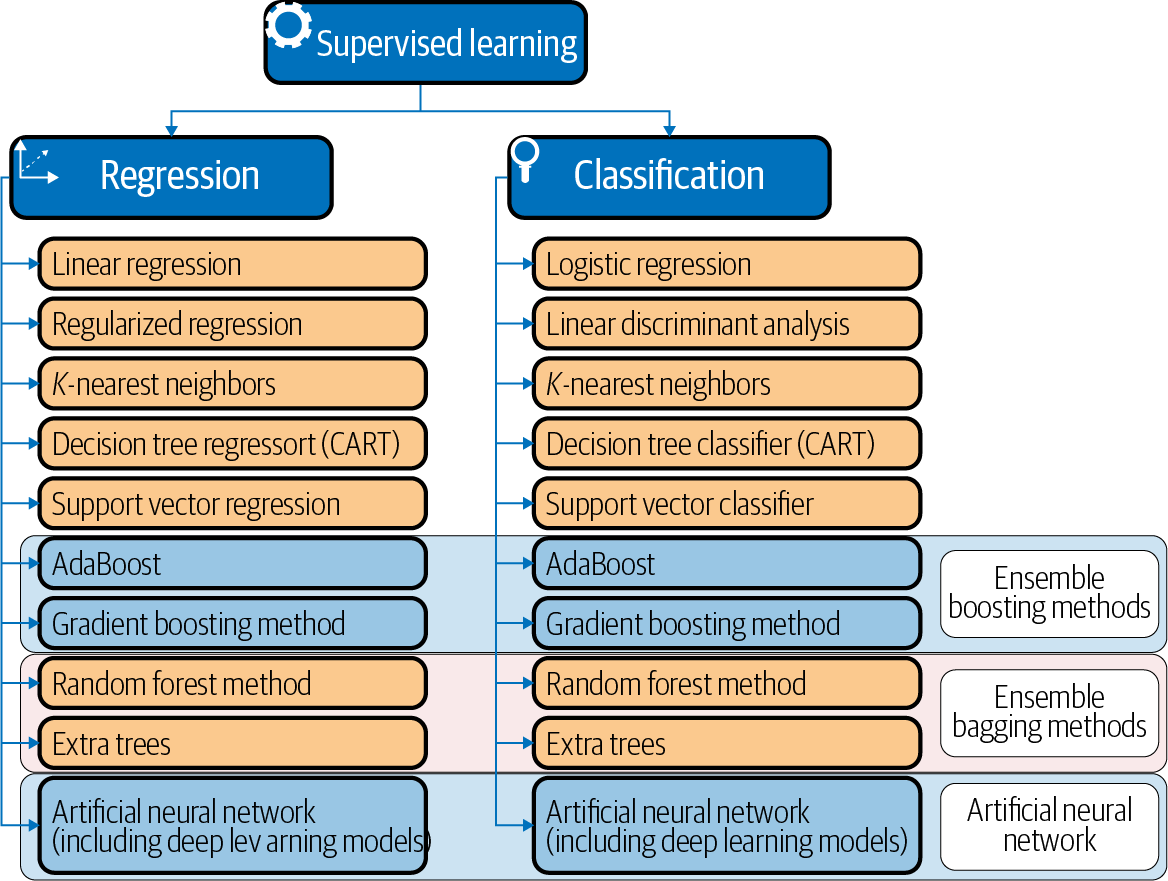
Classification predictive modeling problems are different from regression predictive modeling problems, as classification is the task of predicting a discrete class label and regression is the task of predicting a continuous quantity. However, both share the same concept of utilizing known variables to make predictions, and there is a significant overlap between the two models. Hence, the models for classification and regression are presented together in this chapter. Figure 4-1 summarizes the list of the models commonly used for classification and regression.
Some models can be used for both classification and regression with small modifications. These are K-nearest neighbors, decision trees, support vector, ensemble bagging/boosting methods, and ANNs (including deep neural networks), as shown in Figure 4-1. However, some models, such as linear regression and logistic regression, cannot (or cannot easily) be used for both problem types.
This section contains the following details about the models:
- Theory of the models.
- Implementation in Scikit-learn or Keras.
- Grid search for different models.
- Pros and cons of the models.
Note
In finance, a key focus is on models that extract signals from previously observed data in order to predict future values for the same time series. This family of time series models predicts continuous output and is more aligned with the supervised regression models. Time series models are covered separately in the supervised regression chapter (Chapter 5).
Linear Regression (Ordinary Least Squares)
Linear regression (Ordinary Least Squares Regression or OLS Regression) is perhaps one of the most well-known and best-understood algorithms in statistics and machine learning. Linear regression is a linear model, e.g., a model that assumes a linear relationship between the input variables (x) and the single output variable (y). The goal of linear regression is to train a linear model to predict a new y given a previously unseen x with as little error as possible.
Our model will be a function that predicts y given �1,�2…��:�=�0+�1�1+…+����
where, �0 is called intercept and �1…�� are the coefficient of the regression.
Implementation in Python
fromsklearn.linear_modelimportLinearRegressionmodel=LinearRegression()model.fit(X,Y)
In the following section, we cover the training of a linear regression model and grid search of the model. However, the overall concepts and related approaches are applicable to all other supervised learning models.
Training a model
As we mentioned in Chapter 3, training a model basically means retrieving the model parameters by minimizing the cost (loss) function. The two steps for training a linear regression model are:Define a cost function (or loss function)
Measures how inaccurate the model’s predictions are. The sum of squared residuals (RSS) as defined in Equation 4-1 measures the squared sum of the difference between the actual and predicted value and is the cost function for linear regression.
Equation 4-1. Sum of squared residuals
���=∑�=1���–�0–∑�=1������2
In this equation, �0 is the intercept; �� represents the coefficient; �1,..,�� are the coefficients of the regression; and ��� represents the ��ℎ observation and ��ℎ variable.Find the parameters that minimize loss
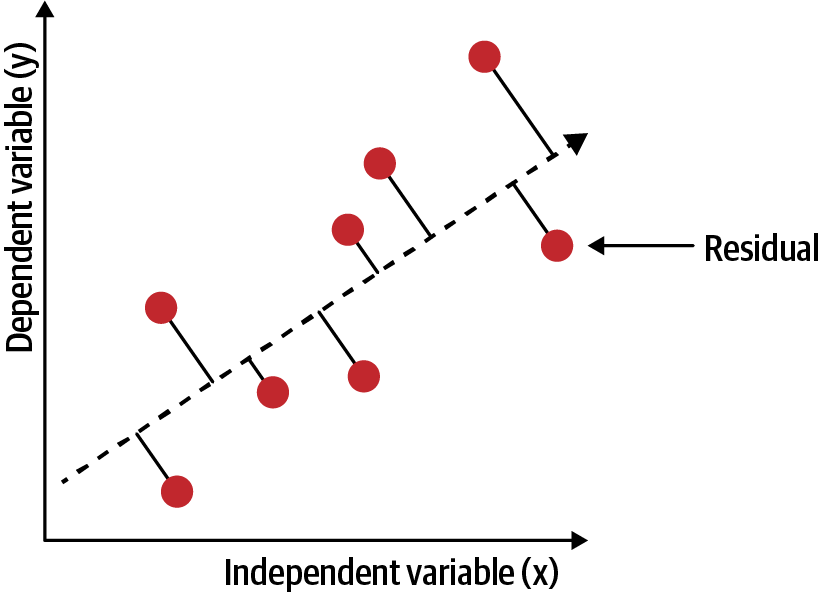
For example, make our model as accurate as possible. Graphically, in two dimensions, this results in a line of best fit as shown in Figure 4-2. In higher dimensions, we would have higher-dimensional hyperplanes. Mathematically, we look at the difference between each real data point (y) and our model’s prediction (ŷ). Square these differences to avoid negative numbers and penalize larger differences, and then add them up and take the average. This is a measure of how well our data fits the line.
Grid search
The overall idea of the grid search is to create a grid of all possible hyperparameter combinations and train the model using each one of them. Hyperparameters are the external characteristic of the model, can be considered the model’s settings, and are not estimated based on data-like model parameters. These hyperparameters are tuned during grid search to achieve better model performance.
Due to its exhaustive search, a grid search is guaranteed to find the optimal parameter within the grid. The drawback is that the size of the grid grows exponentially with the addition of more parameters or more considered values.
The GridSearchCV class in the model_selection module of the sklearn package facilitates the systematic evaluation of all combinations of the hyperparameter values that we would like to test.
The first step is to create a model object. We then define a dictionary where the keywords name the hyperparameters and the values list the parameter settings to be tested. For linear regression, the hyperparameter is fit_intercept, which is a boolean variable that determines whether or not to calculate the intercept for this model. If set to False, no intercept will be used in calculations:
model=LinearRegression()param_grid={'fit_intercept':[True,False]}}
The second step is to instantiate the GridSearchCV object and provide the estimator object and parameter grid, as well as a scoring method and cross validation choice, to the initialization method. Cross validation is a resampling procedure used to evaluate machine learning models, and scoring parameter is the evaluation metrics of the model:1
With all settings in place, we can fit GridSearchCV:
grid=GridSearchCV(estimator=model,param_grid=param_grid,scoring='r2',\cv=kfold)grid_result=grid.fit(X,Y)
Advantages and disadvantages
In terms of advantages, linear regression is easy to understand and interpret. However, it may not work well when there is a nonlinear relationship between predicted and predictor variables. Linear regression is prone to overfitting (which we will discuss in the next section) and when a large number of features are present, it may not handle irrelevant features well. Linear regression also requires the data to follow certain assumptions, such as the absence of multicollinearity. If the assumptions fail, then we cannot trust the results obtained.
Regularized Regression
When a linear regression model contains many independent variables, their coefficients will be poorly determined, and the model will have a tendency to fit extremely well to the training data (data used to build the model) but fit poorly to testing data (data used to test how good the model is). This is known as overfitting or high variance.
One popular technique to control overfitting is regularization, which involves the addition of a penalty term to the error or loss function to discourage the coefficients from reaching large values. Regularization, in simple terms, is a penalty mechanism that applies shrinkage to model parameters (driving them closer to zero) in order to build a model with higher prediction accuracy and interpretation. Regularized regression has two advantages over linear regression:Prediction accuracy
The performance of the model working better on the testing data suggests that the model is trying to generalize from training data. A model with too many parameters might try to fit noise specific to the training data. By shrinking or setting some coefficients to zero, we trade off the ability to fit complex models (higher bias) for a more generalizable model (lower variance).Interpretation
A large number of predictors may complicate the interpretation or communication of the big picture of the results. It may be preferable to sacrifice some detail to limit the model to a smaller subset of parameters with the strongest effects.
The common ways to regularize a linear regression model are as follows:L1 regularization or Lasso regression
Lasso regression performs L1 regularization by adding a factor of the sum of the absolute value of coefficients in the cost function (RSS) for linear regression, as mentioned in Equation 4-1. The equation for lasso regularization can be represented as follows:
������������=���+�*∑�=1���
L1 regularization can lead to zero coefficients (i.e., some of the features are completely neglected for the evaluation of output). The larger the value of �, the more features are shrunk to zero. This can eliminate some features entirely and give us a subset of predictors, reducing model complexity. So Lasso regression not only helps in reducing overfitting, but also can help in feature selection. Predictors not shrunk toward zero signify that they are important, and thus L1 regularization allows for feature selection (sparse selection). The regularization parameter (�) can be controlled, and a lambda value of zero produces the basic linear regression equation.
A lasso regression model can be constructed using the Lasso class of the sklearn package of Python, as shown in the code snippet that follows:
fromsklearn.linear_modelimportLassomodel=Lasso()model.fit(X,Y)
L2 regularization or Ridge regression
Ridge regression performs L2 regularization by adding a factor of the sum of the square of coefficients in the cost function (RSS) for linear regression, as mentioned in Equation 4-1. The equation for ridge regularization can be represented as follows:
������������=���+�*∑�=1���2
Ridge regression puts constraint on the coefficients. The penalty term (�) regularizes the coefficients such that if the coefficients take large values, the optimization function is penalized. So ridge regression shrinks the coefficients and helps to reduce the model complexity. Shrinking the coefficients leads to a lower variance and a lower error value. Therefore, ridge regression decreases the complexity of a model but does not reduce the number of variables; it just shrinks their effect. When � is closer to zero, the cost function becomes similar to the linear regression cost function. So the lower the constraint (low �) on the features, the more the model will resemble the linear regression model.
A ridge regression model can be constructed using the Ridge class of the sklearn package of Python, as shown in the code snippet that follows:
fromsklearn.linear_modelimportRidgemodel=Ridge()model.fit(X,Y)
Elastic net
Elastic nets add regularization terms to the model, which are a combination of both L1 and L2 regularization, as shown in the following equation:
������������=���+�*(1–�)/2*∑�=1���2+�*∑�=1���
In addition to setting and choosing a � value, an elastic net also allows us to tune the alpha parameter, where � = 0 corresponds to ridge and � = 1 to lasso. Therefore, we can choose an � value between 0 and 1 to optimize the elastic net. Effectively, this will shrink some coefficients and set some to 0 for sparse selection.
An elastic net regression model can be constructed using the ElasticNet class of the sklearn package of Python, as shown in the following code snippet:
fromsklearn.linear_modelimportElasticNetmodel=ElasticNet()model.fit(X,Y)
For all the regularized regression, � is the key parameter to tune during grid search in Python. In an elastic net, � can be an additional parameter to tune.
Logistic Regression
Logistic regression is one of the most widely used algorithms for classification. The logistic regression model arises from the desire to model the probabilities of the output classes given a function that is linear in x, at the same time ensuring that output probabilities sum up to one and remain between zero and one as we would expect from probabilities.
If we train a linear regression model on several examples where Y = 0 or 1, we might end up predicting some probabilities that are less than zero or greater than one, which doesn’t make sense. Instead, we use a logistic regression model (or logit model), which is a modification of linear regression that makes sure to output a probability between zero and one by applying the sigmoid function.2
Equation 4-2 shows the equation for a logistic regression model. Similar to linear regression, input values (x) are combined linearly using weights or coefficient values to predict an output value (y). The output coming from Equation 4-2 is a probability that is transformed into a binary value (0 or 1) to get the model prediction.
Equation 4-2. Logistic regression equation
�=exp(�0+�1�1+….+���1)1+exp(�0+�1�1+….+���1)
Where y is the predicted output, �0 is the bias or intercept term and B1 is the coefficient for the single input value (x). Each column in the input data has an associated � coefficient (a constant real value) that must be learned from the training data.
In logistic regression, the cost function is basically a measure of how often we predicted one when the true answer was zero, or vice versa. Training the logistic regression coefficients is done using techniques such as maximum likelihood estimation (MLE) to predict values close to 1 for the default class and close to 0 for the other class.3
A logistic regression model can be constructed using the LogisticRegression class of the sklearn package of Python, as shown in the following code snippet:
fromsklearn.linear_modelimportLogisticRegressionmodel=LogisticRegression()model.fit(X,Y)
Hyperparameters
Regularization (penalty in sklearn)
Similar to linear regression, logistic regression can have regularization, which can be L1, L2, or elasticnet. The values in the sklearn library are [l1, l2, elasticnet].Regularization strength (C in sklearn)
This parameter controls the regularization strength. Good values of the penalty parameters can be [100, 10, 1.0, 0.1, 0.01].
Advantages and disadvantages
In terms of the advantages, the logistic regression model is easy to implement, has good interpretability, and performs very well on linearly separable classes. The output of the model is a probability, which provides more insight and can be used for ranking. The model has small number of hyperparameters. Although there may be risk of overfitting, this may be addressed using L1/L2 regularization, similar to the way we addressed overfitting for the linear regression models.
In terms of disadvantages, the model may overfit when provided with large numbers of features. Logistic regression can only learn linear functions and is less suitable to complex relationships between features and the target variable. Also, it may not handle irrelevant features well, especially if the features are strongly correlated.
Support Vector Machine
The objective of the support vector machine (SVM) algorithm is to maximize the margin (shown as shaded area in Figure 4-3), which is defined as the distance between the separating hyperplane (or decision boundary) and the training samples that are closest to this hyperplane, the so-called support vectors. The margin is calculated as the perpendicular distance from the line to only the closest points, as shown in Figure 4-3. Hence, SVM calculates a maximum-margin boundary that leads to a homogeneous partition of all data points.
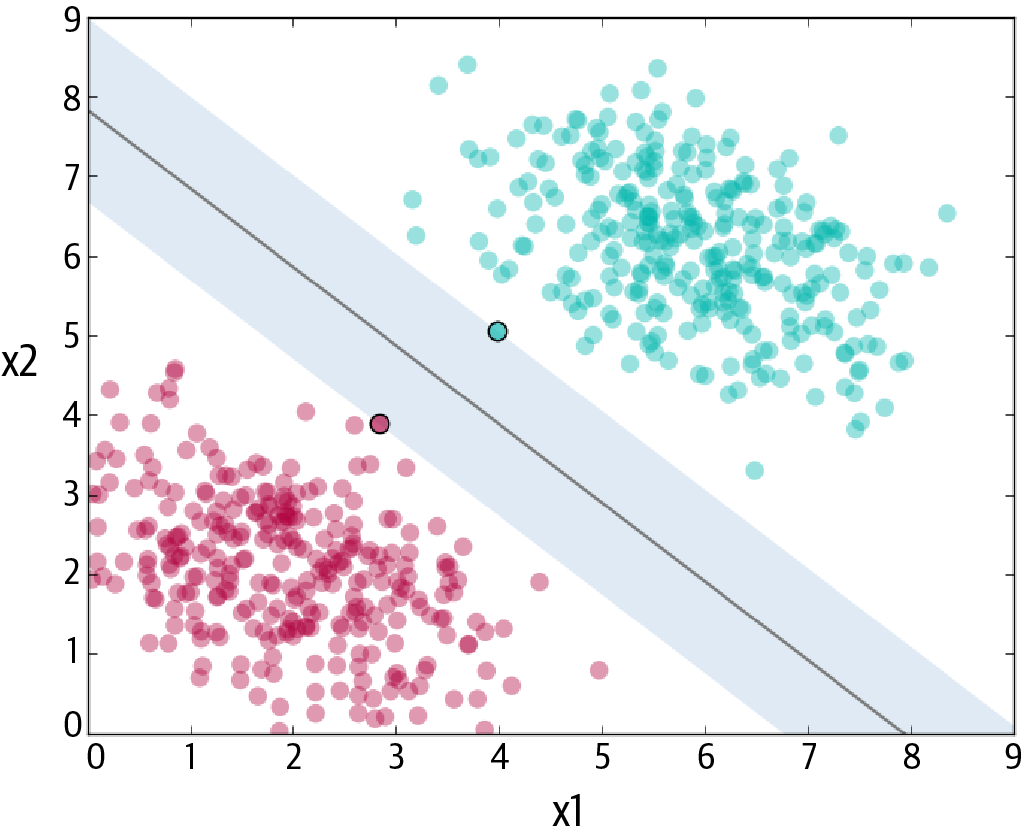
In practice, the data is messy and cannot be separated perfectly with a hyperplane. The constraint of maximizing the margin of the line that separates the classes must be relaxed. This change allows some points in the training data to violate the separating line. An additional set of coefficients is introduced that give the margin wiggle room in each dimension. A tuning parameter is introduced, simply called C, that defines the magnitude of the wiggle allowed across all dimensions. The larger the value of C, the more violations of the hyperplane are permitted.
In some cases, it is not possible to find a hyperplane or a linear decision boundary, and kernels are used. A kernel is just a transformation of the input data that allows the SVM algorithm to treat/process the data more easily. Using kernels, the original data is projected into a higher dimension to classify the data better.
SVM is used for both classification and regression. We achieve this by converting the original optimization problem into a dual problem. For regression, the trick is to reverse the objective. Instead of trying to fit the largest possible street between two classes while limiting margin violations, SVM regression tries to fit as many instances as possible on the street (shaded area in Figure 4-3) while limiting margin violations. The width of the street is controlled by a hyperparameter.
The SVM regression and classification models can be constructed using the sklearn package of Python, as shown in the following code snippets:
Regression
fromsklearn.svmimportSVRmodel=SVR()model.fit(X,Y)
Classification
fromsklearn.svmimportSVCmodel=SVC()model.fit(X,Y)
Hyperparameters
The following key parameters are present in the sklearn implementation of SVM and can be tweaked while performing the grid search:Kernels (kernel in sklearn)
The choice of kernel controls the manner in which the input variables will be projected. There are many kernels to choose from, but linear and RBF are the most common.Penalty (C in sklearn)
The penalty parameter tells the SVM optimization how much you want to avoid misclassifying each training example. For large values of the penalty parameter, the optimization will choose a smaller-margin hyperplane. Good values might be a log scale from 10 to 1,000.
Advantages and disadvantages
In terms of advantages, SVM is fairly robust against overfitting, especially in higher dimensional space. It handles the nonlinear relationships quite well, with many kernels to choose from. Also, there is no distributional requirement for the data.
In terms of disadvantages, SVM can be inefficient to train and memory-intensive to run and tune. It doesn’t perform well with large datasets. It requires the feature scaling of the data. There are also many hyperparameters, and their meanings are often not intuitive.
K-Nearest Neighbors
K-nearest neighbors (KNN) is considered a “lazy learner,” as there is no learning required in the model. For a new data point, predictions are made by searching through the entire training set for the K most similar instances (the neighbors) and summarizing the output variable for those K instances.
To determine which of the K instances in the training dataset are most similar to a new input, a distance measure is used. The most popular distance measure is Euclidean distance, which is calculated as the square root of the sum of the squared differences between a point a and a point b across all input attributes i, and which is represented as �(�,�)=∑�=1�(��–��)2. Euclidean distance is a good distance measure to use if the input variables are similar in type.
Another distance metric is Manhattan distance, in which the distance between point a and point b is represented as �(�,�)=∑�=1�|��–��|. Manhattan distance is a good measure to use if the input variables are not similar in type.
The steps of KNN can be summarized as follows:
- Choose the number of K and a distance metric.
- Find the K-nearest neighbors of the sample that we want to classify.
- Assign the class label by majority vote.
KNN regression and classification models can be constructed using the sklearn package of Python, as shown in the following code:
Classification
fromsklearn.neighborsimportKNeighborsClassifiermodel=KNeighborsClassifier()model.fit(X,Y)
Regression
fromsklearn.neighborsimportKNeighborsRegressormodel=KNeighborsRegressor()model.fit(X,Y)
Hyperparameters
The following key parameters are present in the sklearn implementation of KNN and can be tweaked while performing the grid search:Number of neighbors (n_neighbors in sklearn)
The most important hyperparameter for KNN is the number of neighbors (n_neighbors). Good values are between 1 and 20.Distance metric (metric in sklearn)
It may also be interesting to test different distance metrics for choosing the composition of the neighborhood. Good values are euclidean and manhattan.
Advantages and disadvantages
In terms of advantages, no training is involved and hence there is no learning phase. Since the algorithm requires no training before making predictions, new data can be added seamlessly without impacting the accuracy of the algorithm. It is intuitive and easy to understand. The model naturally handles multiclass classification and can learn complex decision boundaries. KNN is effective if the training data is large. It is also robust to noisy data, and there is no need to filter the outliers.
In terms of the disadvantages, the distance metric to choose is not obvious and difficult to justify in many cases. KNN performs poorly on high dimensional datasets. It is expensive and slow to predict new instances because the distance to all neighbors must be recalculated. KNN is sensitive to noise in the dataset. We need to manually input missing values and remove outliers. Also, feature scaling (standardization and normalization) is required before applying the KNN algorithm to any dataset; otherwise, KNN may generate wrong predictions.
Linear Discriminant Analysis
The objective of the linear discriminant analysis (LDA) algorithm is to project the data onto a lower-dimensional space in a way that the class separability is maximized and the variance within a class is minimized.4
During the training of the LDA model, the statistical properties (i.e., mean and covariance matrix) of each class are computed. The statistical properties are estimated on the basis of the following assumptions about the data:
- Data is normally distributed, so that each variable is shaped like a bell curve when plotted.
- Each attribute has the same variance, and the values of each variable vary around the mean by the same amount on average.
To make a prediction, LDA estimates the probability that a new set of inputs belongs to every class. The output class is the one that has the highest probability.
Implementation in Python and hyperparameters
The LDA classification model can be constructed using the sklearn package of Python, as shown in the following code snippet:
fromsklearn.discriminant_analysisimportLinearDiscriminantAnalysismodel=LinearDiscriminantAnalysis()model.fit(X,Y)
The key hyperparameter for the LDA model is number of components for dimensionality reduction, which is represented by n_components in sklearn.
Advantages and disadvantages
In terms of advantages, LDA is a relatively simple model with fast implementation and is easy to implement. In terms of disadvantages, it requires feature scaling and involves complex matrix operations.
Classification and Regression Trees
In the most general terms, the purpose of an analysis via tree-building algorithms is to determine a set of if–then logical (split) conditions that permit accurate prediction or classification of cases. Classification and regression trees (or CART or decision tree classifiers) are attractive models if we care about interpretability. We can think of this model as breaking down our data and making a decision based on asking a series of questions. This algorithm is the foundation of ensemble methods such as random forest and gradient boosting method.
Representation
The model can be represented by a binary tree (or decision tree), where each node is an input variable x with a split point and each leaf contains an output variable y for prediction.
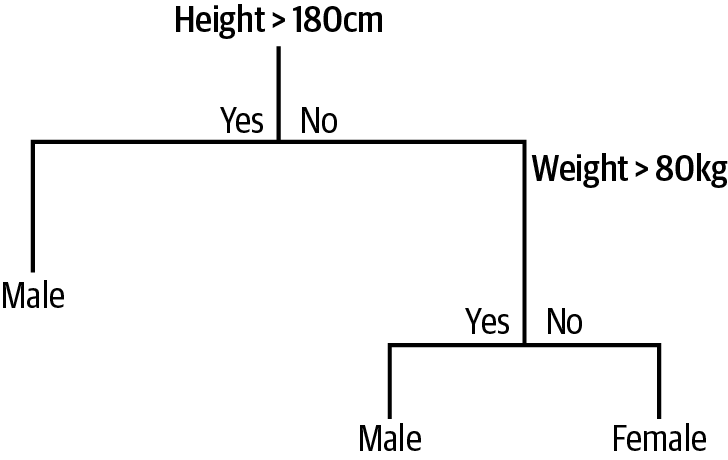
Figure 4-4 shows an example of a simple classification tree to predict whether a person is a male or a female based on two inputs of height (in centimeters) and weight (in kilograms).
Learning a CART model
Creating a binary tree is actually a process of dividing up the input space. A greedy approach called recursive binary splitting is used to divide the space. This is a numerical procedure in which all the values are lined up and different split points are tried and tested using a cost (loss) function. The split with the best cost (lowest cost, because we minimize cost) is selected. All input variables and all possible split points are evaluated and chosen in a greedy manner (e.g., the very best split point is chosen each time).
For regression predictive modeling problems, the cost function that is minimized to choose split points is the sum of squared errors across all training samples that fall within the rectangle:∑�=1�(��–�����������)2
where �� is the output for the training sample and prediction is the predicted output for the rectangle. For classification, the Gini cost function is used; it provides an indication of how pure the leaf nodes are (i.e., how mixed the training data assigned to each node is) and is defined as:�=∑�=1���*(1–��)
where G is the Gini cost over all classes and �� is the number of training instances with class k in the rectangle of interest. A node that has all classes of the same type (perfect class purity) will have G = 0, while a node that has a 50–50 split of classes for a binary classification problem (worst purity) will have G = 0.5.
Stopping criterion
The recursive binary splitting procedure described in the preceding section needs to know when to stop splitting as it works its way down the tree with the training data. The most common stopping procedure is to use a minimum count on the number of training instances assigned to each leaf node. If the count is less than some minimum, then the split is not accepted and the node is taken as a final leaf node.
Pruning the tree
The stopping criterion is important as it strongly influences the performance of the tree. Pruning can be used after learning the tree to further lift performance. The complexity of a decision tree is defined as the number of splits in the tree. Simpler trees are preferred as they are faster to run and easy to understand, consume less memory during processing and storage, and are less likely to overfit the data. The fastest and simplest pruning method is to work through each leaf node in the tree and evaluate the effect of removing it using a test set. A leaf node is removed only if doing so results in a drop in the overall cost function on the entire test set. The removal of nodes can be stopped when no further improvements can be made.
Implementation in Python
CART regression and classification models can be constructed using the sklearn package of Python, as shown in the following code snippet:
Classification
fromsklearn.treeimportDecisionTreeClassifiermodel=DecisionTreeClassifier()model.fit(X,Y)
Regression
fromsklearn.treeimportDecisionTreeRegressormodel=DecisionTreeRegressor()model.fit(X,Y)
Hyperparameters
CART has many hyperparameters. However, the key hyperparameter is the maximum depth of the tree model, which is the number of components for dimensionality reduction, and which is represented by max_depth in the sklearn package. Good values can range from 2 to 30 depending on the number of features in the data.
Advantages and disadvantages
In terms of advantages, CART is easy to interpret and can adapt to learn complex relationships. It requires little data preparation, and data typically does not need to be scaled. Feature importance is built in due to the way decision nodes are built. It performs well on large datasets. It works for both regression and classification problems.
In terms of disadvantages, CART is prone to overfitting unless pruning is used. It can be very nonrobust, meaning that small changes in the training dataset can lead to quite major differences in the hypothesis function that gets learned. CART generally has worse performance than ensemble models, which are covered next.
Ensemble Models
The goal of ensemble models is to combine different classifiers into a meta-classifier that has better generalization performance than each individual classifier alone. For example, assuming that we collected predictions from 10 experts, ensemble methods would allow us to strategically combine their predictions to come up with a prediction that is more accurate and robust than the experts’ individual predictions.
The two most popular ensemble methods are bagging and boosting. Bagging (or bootstrap aggregation) is an ensemble technique of training several individual models in a parallel way. Each model is trained by a random subset of the data. Boosting, on the other hand, is an ensemble technique of training several individual models in a sequential way. This is done by building a model from the training data and then creating a second model that attempts to correct the errors of the first model. Models are added until the training set is predicted perfectly or a maximum number of models is added. Each individual model learns from mistakes made by the previous model. Just like the decision trees themselves, bagging and boosting can be used for classification and regression problems.
By combining individual models, the ensemble model tends to be more flexible (less bias) and less data-sensitive (less variance).5 Ensemble methods combine multiple, simpler algorithms to obtain better performance.
In this section we will cover random forest, AdaBoost, the gradient boosting method, and extra trees, along with their implementation using sklearn package.
Random forest
Random forest is a tweaked version of bagged decision trees. In order to understand a random forest algorithm, let us first understand the bagging algorithm. Assuming we have a dataset of one thousand instances, the steps of bagging are:
- Create many (e.g., one hundred) random subsamples of our dataset.
- Train a CART model on each sample.
- Given a new dataset, calculate the average prediction from each model and aggregate the prediction by each tree to assign the final label by majority vote.
A problem with decision trees like CART is that they are greedy. They choose the variable to split by using a greedy algorithm that minimizes error. Even after bagging, the decision trees can have a lot of structural similarities and result in high correlation in their predictions. Combining predictions from multiple models in ensembles works better if the predictions from the submodels are uncorrelated, or at best are weakly correlated. Random forest changes the learning algorithm in such a way that the resulting predictions from all of the subtrees have less correlation.
In CART, when selecting a split point, the learning algorithm is allowed to look through all variables and all variable values in order to select the most optimal split point. The random forest algorithm changes this procedure such that each subtree can access only a random sample of features when selecting the split points. The number of features that can be searched at each split point (m) must be specified as a parameter to the algorithm.
As the bagged decision trees are constructed, we can calculate how much the error function drops for a variable at each split point. In regression problems, this may be the drop in sum squared error, and in classification, this might be the Gini cost. The bagged method can provide feature importance by calculating and averaging the error function drop for individual variables.
Implementation in Python
Random forest regression and classification models can be constructed using the sklearn package of Python, as shown in the following code:
Classification
fromsklearn.ensembleimportRandomForestClassifiermodel=RandomForestClassifier()model.fit(X,Y)
Regression
fromsklearn.ensembleimportRandomForestRegressormodel=RandomForestRegressor()model.fit(X,Y)
Hyperparameters
Some of the main hyperparameters that are present in the sklearn implementation of random forest and that can be tweaked while performing the grid search are:Maximum number of features (max_features in sklearn)
This is the most important parameter. It is the number of random features to sample at each split point. You could try a range of integer values, such as 1 to 20, or 1 to half the number of input features.Number of estimators (n_estimators in sklearn)
This parameter represents the number of trees. Ideally, this should be increased until no further improvement is seen in the model. Good values might be a log scale from 10 to 1,000.
Advantages and disadvantages
The random forest algorithm (or model) has gained huge popularity in ML applications during the last decade due to its good performance, scalability, and ease of use. It is flexible and naturally assigns feature importance scores, so it can handle redundant feature columns. It scales to large datasets and is generally robust to overfitting. The algorithm doesn’t need the data to be scaled and can model a nonlinear relationship.
In terms of disadvantages, random forest can feel like a black box approach, as we have very little control over what the model does, and the results may be difficult to interpret. Although random forest does a good job at classification, it may not be good for regression problems, as it does not give a precise continuous nature prediction. In the case of regression, it doesn’t predict beyond the range in the training data and may overfit datasets that are particularly noisy.
Extra trees
Extra trees, otherwise known as extremely randomized trees, is a variant of a random forest; it builds multiple trees and splits nodes using random subsets of features similar to random forest. However, unlike random forest, where observations are drawn with replacement, the observations are drawn without replacement in extra trees. So there is no repetition of observations.
Additionally, random forest selects the best split to convert the parent into the two most homogeneous child nodes.6 However, extra trees selects a random split to divide the parent node into two random child nodes. In extra trees, randomness doesn’t come from bootstrapping the data; it comes from the random splits of all observations.
In real-world cases, performance is comparable to an ordinary random forest, sometimes a bit better. The advantages and disadvantages of extra trees are similar to those of random forest.
Implementation in Python
Extra trees regression and classification models can be constructed using the sklearn package of Python, as shown in the following code snippet. The hyperparameters of extra trees are similar to random forest, as shown in the previous section:
Classification
fromsklearn.ensembleimportExtraTreesClassifiermodel=ExtraTreesClassifier()model.fit(X,Y)
Regression
fromsklearn.ensembleimportExtraTreesRegressormodel=ExtraTreesRegressor()model.fit(X,Y)
Adaptive Boosting (AdaBoost)
Adaptive Boosting or AdaBoost is a boosting technique in which the basic idea is to try predictors sequentially, and each subsequent model attempts to fix the errors of its predecessor. At each iteration, the AdaBoost algorithm changes the sample distribution by modifying the weights attached to each of the instances. It increases the weights of the wrongly predicted instances and decreases the ones of the correctly predicted instances.
The steps of the AdaBoost algorithm are:
- Initially, all observations are given equal weights.
- A model is built on a subset of data, and using this model, predictions are made on the whole dataset. Errors are calculated by comparing the predictions and actual values.
- While creating the next model, higher weights are given to the data points that were predicted incorrectly. Weights can be determined using the error value. For instance, the higher the error, the more weight is assigned to the observation.
- This process is repeated until the error function does not change, or until the maximum limit of the number of estimators is reached.
Implementation in Python
AdaBoost regression and classification models can be constructed using the sklearn package of Python, as shown in the following code snippet:
Classification
fromsklearn.ensembleimportAdaBoostClassifiermodel=AdaBoostClassifier()model.fit(X,Y)
Regression
fromsklearn.ensembleimportAdaBoostRegressormodel=AdaBoostRegressor()model.fit(X,Y)
Hyperparameters
Some of the main hyperparameters that are present in the sklearn implementation of AdaBoost and that can be tweaked while performing the grid search are as follows:Learning rate (learning_rate in sklearn)
Learning rate shrinks the contribution of each classifier/regressor. It can be considered on a log scale. The sample values for grid search can be 0.001, 0.01, and 0.1.Number of estimators (n_estimators in sklearn)
This parameter represents the number of trees. Ideally, this should be increased until no further improvement is seen in the model. Good values might be a log scale from 10 to 1,000.
Advantages and disadvantages
In terms of advantages, AdaBoost has a high degree of precision. AdaBoost can achieve similar results to other models with much less tweaking of parameters or settings. The algorithm doesn’t need the data to be scaled and can model a nonlinear relationship.
In terms of disadvantages, the training of AdaBoost is time consuming. AdaBoost can be sensitive to noisy data and outliers, and data imbalance leads to a decrease in classification accuracy
Gradient boosting method
Gradient boosting method (GBM) is another boosting technique similar to AdaBoost, where the general idea is to try predictors sequentially. Gradient boosting works by sequentially adding the previous underfitted predictions to the ensemble, ensuring the errors made previously are corrected.
The following are the steps of the gradient boosting algorithm:
- A model (which can be referred to as the first weak learner) is built on a subset of data. Using this model, predictions are made on the whole dataset.
- Errors are calculated by comparing the predictions and actual values, and the loss is calculated using the loss function.
- A new model is created using the errors of the previous step as the target variable. The objective is to find the best split in the data to minimize the error. The predictions made by this new model are combined with the predictions of the previous. New errors are calculated using this predicted value and actual value.
- This process is repeated until the error function does not change or until the maximum limit of the number of estimators is reached.
Contrary to AdaBoost, which tweaks the instance weights at every interaction, this method tries to fit the new predictor to the residual errors made by the previous predictor.
Implementation in Python and hyperparameters
Gradient boosting method regression and classification models can be constructed using the sklearn package of Python, as shown in the following code snippet. The hyperparameters of gradient boosting method are similar to AdaBoost, as shown in the previous section:
Classification
fromsklearn.ensembleimportGradientBoostingClassifiermodel=GradientBoostingClassifier()model.fit(X,Y)
Regression
fromsklearn.ensembleimportGradientBoostingRegressormodel=GradientBoostingRegressor()model.fit(X,Y)
Advantages and disadvantages
In terms of advantages, gradient boosting method is robust to missing data, highly correlated features, and irrelevant features in the same way as random forest. It naturally assigns feature importance scores, with slightly better performance than random forest. The algorithm doesn’t need the data to be scaled and can model a nonlinear relationship.
In terms of disadvantages, it may be more prone to overfitting than random forest, as the main purpose of the boosting approach is to reduce bias and not variance. It has many hyperparameters to tune, so model development may not be as fast. Also, feature importance may not be robust to variation in the training dataset.
ANN-Based Models
In Chapter 3 we covered the basics of ANNs, along with the architecture of ANNs and their training and implementation in Python. The details provided in that chapter are applicable across all areas of machine learning, including supervised learning. However, there are a few additional details from the supervised learning perspective, which we will cover in this section.
Neural networks are reducible to a classification or regression model with the activation function of the node in the output layer. In the case of a regression problem, the output node has linear activation function (or no activation function). A linear function produces a continuous output ranging from -inf to +inf. Hence, the output layer will be the linear function of the nodes in the layer before the output layer, and it will be a regression-based model.
In the case of a classification problem, the output node has a sigmoid or softmax activation function. A sigmoid or softmax function produces an output ranging from zero to one to represent the probability of target value. Softmax function can also be used for multiple groups for classification.
ANN using sklearn
ANN regression and classification models can be constructed using the sklearn package of Python, as shown in the following code snippet:
Classification
fromsklearn.neural_networkimportMLPClassifiermodel=MLPClassifier()model.fit(X,Y)
Regression
fromsklearn.neural_networkimportMLPRegressormodel=MLPRegressor()model.fit(X,Y)
Hyperparameters
As we saw in Chapter 3, ANN has many hyperparameters. Some of the hyperparameters that are present in the sklearn implementation of ANN and can be tweaked while performing the grid search are:Hidden Layers (hidden_layer_sizes in sklearn)
It represents the number of layers and nodes in the ANN architecture. In sklearn implementation of ANN, the ith element represents the number of neurons in the ith hidden layer. A sample value for grid search in the sklearn implementation can be [(20,), (50,), (20, 20), (20, 30, 20)].Activation Function (activation in sklearn)
It represents the activation function of a hidden layer. Some of the activation functions defined in Chapter 3, such as sigmoid, relu, or tanh, can be used.
Deep neural network
ANNs with more than a single hidden layer are often called deep networks. We prefer using the library Keras to implement such networks, given the flexibility of the library. The detailed implementation of a deep neural network in Keras was shown in Chapter 3. Similar to MLPClassifier and MLPRegressor in sklearn for classification and regression, Keras has modules called KerasClassifier and KerasRegressor that can be used for creating classification and regression models with deep network.
A popular problem in finance is time series prediction, which is predicting the next value of a time series based on a historical overview. Some of the deep neural networks, such as recurrent neural network (RNN), can be directly used for time series prediction. The details of this approach are provided in Chapter 5.
Advantages and disadvantages
The main advantage of an ANN is that it captures the nonlinear relationship between the variables quite well. ANN can more easily learn rich representations and is good with a large number of input features with a large dataset. ANN is flexible in how it can be used. This is evident from its use across a wide variety of areas in machine learning and AI, including reinforcement learning and NLP, as discussed in Chapter 3.
The main disadvantage of ANN is the interpretability of the model, which is a drawback that often cannot be ignored and is sometimes the determining factor when choosing a model. ANN is not good with small datasets and requires a lot of tweaking and guesswork. Choosing the right topology/algorithms to solve a problem is difficult. Also, ANN is computationally expensive and can take a lot of time to train.
Using ANNs for supervised learning in finance
If a simple model such as linear or logistic regression perfectly fits your problem, don’t bother with ANN. However, if you are modeling a complex dataset and feel a need for better prediction power, give ANN a try. ANN is one of the most flexible models in adapting itself to the shape of the data, and using it for supervised learning problems can be an interesting and valuable exercise.
Model Performance
In the previous section, we discussed grid search as a way to find the right hyperparameter to achieve better performance. In this section, we will expand on that process by discussing the key components of evaluating the model performance, which are overfitting, cross validation, and evaluation metrics.
Overfitting and Underfitting
A common problem in machine learning is overfitting, which is defined by learning a function that perfectly explains the training data that the model learned from but doesn’t generalize well to unseen test data. Overfitting happens when a model overlearns from the training data to the point that it starts picking up idiosyncrasies that aren’t representative of patterns in the real world. This becomes especially problematic as we make our models increasingly more complex. Underfitting is a related issue in which the model is not complex enough to capture the underlying trend in the data. Figure 4-5 illustrates overfitting and underfitting. The left-hand panel of Figure 4-5 shows a linear regression model; a straight line clearly underfits the true function. The middle panel shows that a high degree polynomial approximates the true relationship reasonably well. On the other hand, a polynomial of a very high degree fits the small sample almost perfectly, and performs best on the training data, but this doesn’t generalize, and it would do a horrible job at explaining a new data point.
The concepts of overfitting and underfitting are closely linked to bias-variance trade-off. Bias refers to the error due to overly simplistic assumptions or faulty assumptions in the learning algorithm. Bias results in underfitting of the data, as shown in the left-hand panel of Figure 4-5. A high bias means our learning algorithm is missing important trends among the features. Variance refers to the error due to an overly complex model that tries to fit the training data as closely as possible. In high variance cases, the model’s predicted values are extremely close to the actual values from the training set. High variance gives rise to overfitting, as shown in the right-hand panel of Figure 4-5. Ultimately, in order to have a good model, we need low bias and low variance.
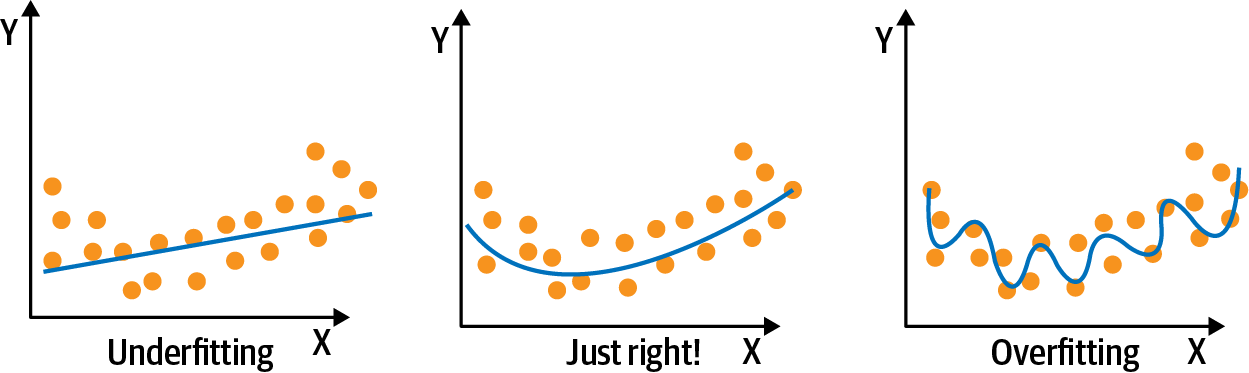
There can be two ways to combat overfitting:Using more training data
The more training data we have, the harder it is to overfit the data by learning too much from any single training example.Using regularization
Adding a penalty in the loss function for building a model that assigns too much explanatory power to any one feature, or allows too many features to be taken into account.
The concept of overfitting and the ways to combat it are applicable across all the supervised learning models. For example, regularized regressions address overfitting in linear regression, as discussed earlier in this chapter.
Cross Validation
One of the challenges of machine learning is training models that are able to generalize well to unseen data (overfitting versus underfitting or a bias-variance trade-off). The main idea behind cross validation is to split the data one time or several times so that each split is used once as a validation set and the remainder is used as a training set: part of the data (the training sample) is used to train the algorithm, and the remaining part (the validation sample) is used for estimating the risk of the algorithm. Cross validation allows us to obtain reliable estimates of the model’s generalization error. It is easiest to understand it with an example. When doing k-fold cross validation, we randomly split the training data into k folds. Then we train the model using k-1 folds and evaluate the performance on the kth fold. We repeat this process k times and average the resulting scores.
Figure 4-6 shows an example of cross validation, where the data is split into five sets and in each round one of the sets is used for validation.
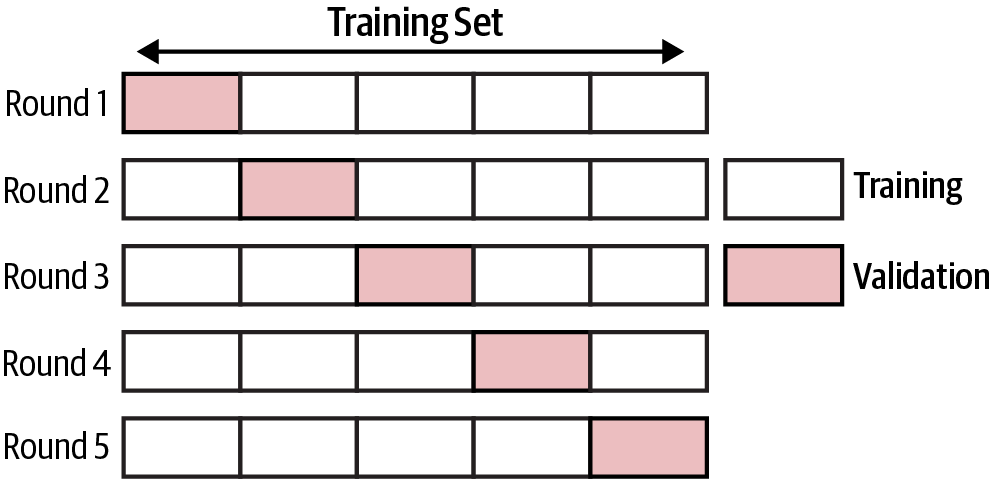
A potential drawback of cross validation is the computational cost, especially when paired with a grid search for hyperparameter tuning. Cross validation can be performed in a couple of lines using the sklearn package; we will perform cross validation in the supervised learning case studies.
In the next section, we cover the evaluation metrics for the supervised learning models that are used to measure and compare the models’ performance.
Evaluation Metrics
The metrics used to evaluate the machine learning algorithms are very important. The choice of metrics to use influences how the performance of machine learning algorithms is measured and compared. The metrics influence both how you weight the importance of different characteristics in the results and your ultimate choice of algorithm.
The main evaluation metrics for regression and classification are illustrated in Figure 4-7.
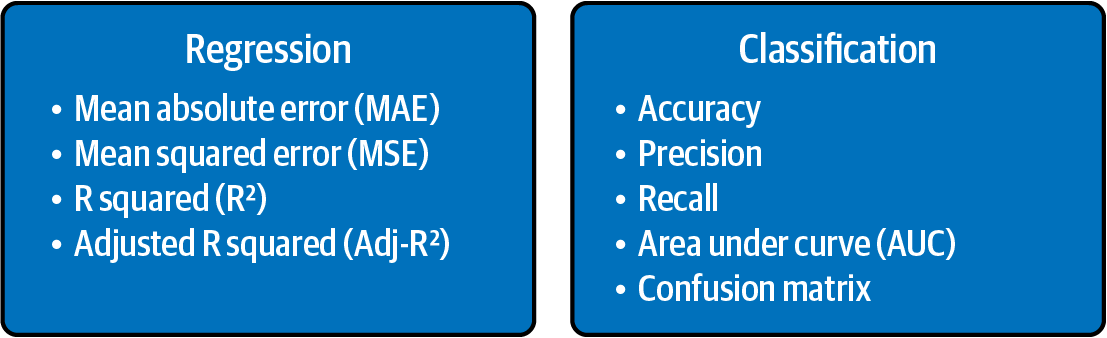
Let us first look at the evaluation metrics for supervised regression.
Mean absolute error
The mean absolute error (MAE) is the sum of the absolute differences between predictions and actual values. The MAE is a linear score, which means that all the individual differences are weighted equally in the average. It gives an idea of how wrong the predictions were. The measure gives an idea of the magnitude of the error, but no idea of the direction (e.g., over- or underpredicting).
Mean squared error
The mean squared error (MSE) represents the sample standard deviation of the differences between predicted values and observed values (called residuals). This is much like the mean absolute error in that it provides a gross idea of the magnitude of the error. Taking the square root of the mean squared error converts the units back to the original units of the output variable and can be meaningful for description and presentation. This is called the root mean squared error (RMSE).
R² metric
The R² metric provides an indication of the “goodness of fit” of the predictions to actual value. In statistical literature this measure is called the coefficient of determination. This is a value between zero and one, for no-fit and perfect fit, respectively.
Adjusted R² metric
Just like R², adjusted R² also shows how well terms fit a curve or line but adjusts for the number of terms in a model. It is given in the following formula:����2=1–(1–�2)(�–1))�–�–1
where n is the total number of observations and k is the number of predictors. Adjusted R² will always be less than or equal to R².
Selecting an evaluation metric for supervised regression
In terms of a preference among these evaluation metrics, if the main goal is predictive accuracy, then RMSE is best. It is computationally simple and is easily differentiable. The loss is symmetric, but larger errors weigh more in the calculation. The MAEs are symmetric but do not weigh larger errors more. R² and adjusted R² are often used for explanatory purposes by indicating how well the selected independent variable(s) explains the variability in the dependent variable(s).
Let us first look at the evaluation metrics for supervised classification.
Classification
For simplicity, we will mostly discuss things in terms of a binary classification problem (i.e., only two outcomes, such as true or false); some common terms are:True positives (TP)
Predicted positive and are actually positive.False positives (FP)
Predicted positive and are actually negative.True negatives (TN)
Predicted negative and are actually negative.False negatives (FN)
Predicted negative and are actually positive.
The difference between three commonly used evaluation metrics for classification, accuracy, precision, and recall, is illustrated in Figure 4-8.

Accuracy
As shown in Figure 4-8, accuracy is the number of correct predictions made as a ratio of all predictions made. This is the most common evaluation metric for classification problems and is also the most misused. It is most suitable when there are an equal number of observations in each class (which is rarely the case) and when all predictions and the related prediction errors are equally important, which is often not the case.
Precision
Precision is the percentage of positive instances out of the total predicted positive instances. Here, the denominator is the model prediction done as positive from the whole given dataset. Precision is a good measure to determine when the cost of false positives is high (e.g., email spam detection).
Recall
Recall (or sensitivity or true positive rate) is the percentage of positive instances out of the total actual positive instances. Therefore, the denominator (true positive + false negative) is the actual number of positive instances present in the dataset. Recall is a good measure when there is a high cost associated with false negatives (e.g., fraud detection).
In addition to accuracy, precision, and recall, some of the other commonly used evaluation metrics for classification are discussed in the following sections.
Area under ROC curve
Area under ROC curve (AUC) is an evaluation metric for binary classification problems. ROC is a probability curve, and AUC represents degree or measure of separability. It tells how much the model is capable of distinguishing between classes. The higher the AUC, the better the model is at predicting zeros as zeros and ones as ones. An AUC of 0.5 means that the model has no class separation capacity whatsoever. The probabilistic interpretation of the AUC score is that if you randomly choose a positive case and a negative case, the probability that the positive case outranks the negative case according to the classifier is given by the AUC.
Confusion matrix
A confusion matrix lays out the performance of a learning algorithm. The confusion matrix is simply a square matrix that reports the counts of the true positive (TP), true negative (TN), false positive (FP), and false negative (FN) predictions of a classifier, as shown in Figure 4-9.
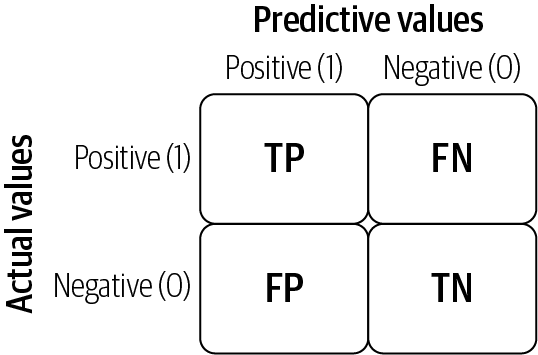
The confusion matrix is a handy presentation of the accuracy of a model with two or more classes. The table presents predictions on the x-axis and accuracy outcomes on the y-axis. The cells of the table are the number of predictions made by the model. For example, a model can predict zero or one, and each prediction may actually have been a zero or a one. Predictions for zero that were actually zero appear in the cell for prediction = 0 and actual = 0, whereas predictions for zero that were actually one appear in the cell for prediction = 0 and actual = 1.
Selecting an evaluation metric for supervised classification
The evaluation metric for classification depends heavily on the task at hand. For example, recall is a good measure when there is a high cost associated with false negatives such as fraud detection. We will further examine these evaluation metrics in the case studies.
Model Selection
Selecting the perfect machine learning model is both an art and a science. Looking at machine learning models, there is no one solution or approach that fits all. There are several factors that can affect your choice of a machine learning model. The main criteria in most of the cases is the model performance that we discussed in the previous section. However, there are many other factors to consider while performing model selection. In the following section, we will go over all such factors, followed by a discussion of model trade-offs.
Factors for Model Selection
The factors considered for the model selection process are as follows:Simplicity
The degree of simplicity of the model. Simplicity usually results in quicker, more scalable, and easier to understand models and results.Training time
Speed, performance, memory usage and overall time taken for model training.Handle nonlinearity in the data
The ability of the model to handle the nonlinear relationship between the variables.Robustness to overfitting
The ability of the model to handle overfitting.Size of the dataset
The ability of the model to handle large number of training examples in the dataset.Number of features
The ability of the model to handle high dimensionality of the feature space.Model interpretation
How explainable is the model? Model interpretability is important because it allows us to take concrete actions to solve the underlying problem.Feature scaling
Does the model require variables to be scaled or normally distributed?
Figure 4-10 compares the supervised learning models on the factors mentioned previously and outlines a general rule-of-thumb to narrow down the search for the best machine learning algorithm7 for a given problem. The table is based on the advantages and disadvantages of different models discussed in the individual model section in this chapter.
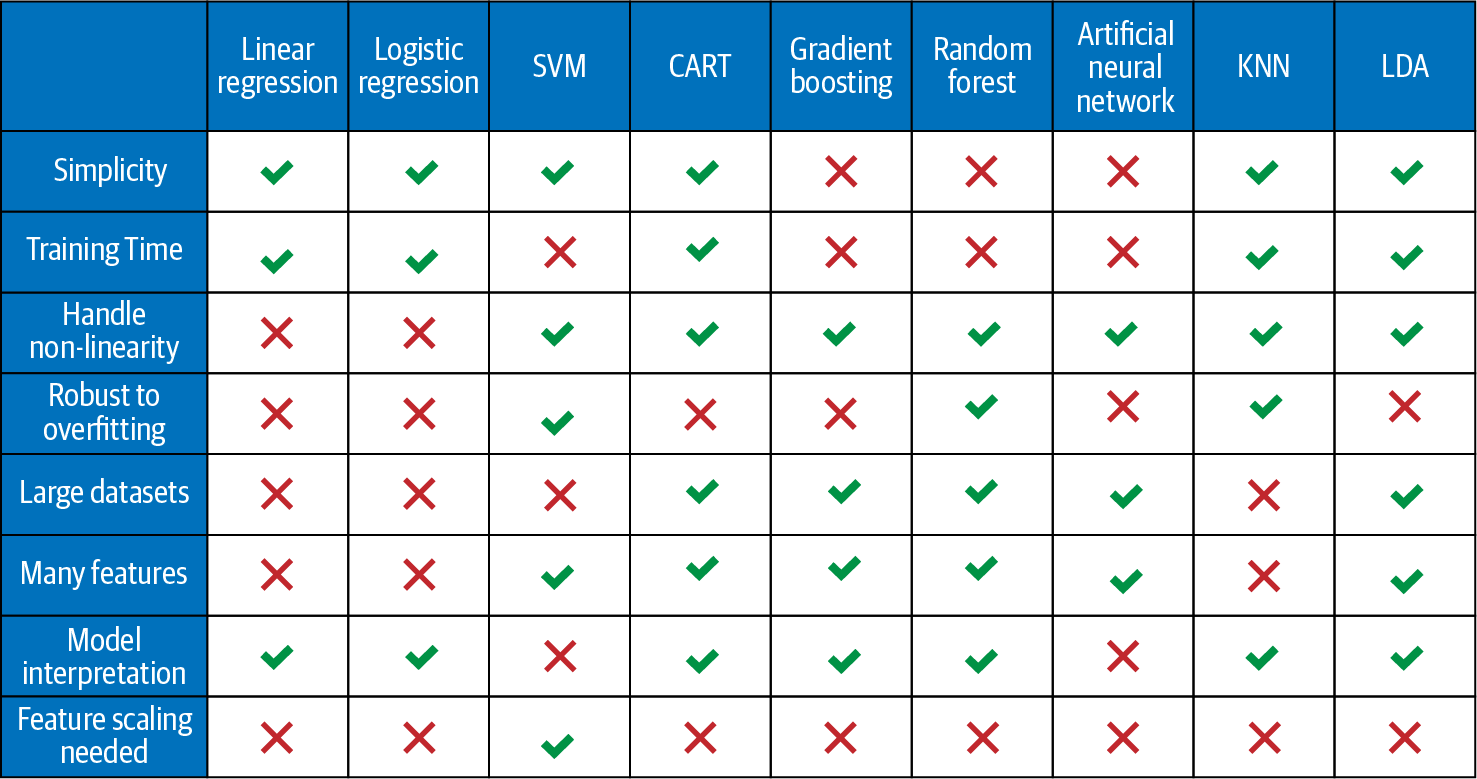
We can see from the table that relatively simple models include linear and logistic regression and as we move towards the ensemble and ANN, the complexity increases. In terms of the training time, the linear models and CART are relatively faster to train as compared to ensemble methods and ANN.
Linear and logistic regression can’t handle nonlinear relationships, while all other models can. SVM can handle the nonlinear relationship between dependent and independent variables with nonlinear kernels.
SVM and random forest tend to overfit less as compared to the linear regression, logistic regression, gradient boosting, and ANN. The degree of overfitting also depends on other parameters, such as size of the data and model tuning, and can be checked by looking at the results of the test set for each model. Also, the boosting methods such as gradient boosting have higher overfitting risk compared to the bagging methods, such as random forest. Recall the focus of gradient boosting is to minimize the bias and not variance.
Linear and logistic regressions are not able to handle large datasets and large number of features well. However, CART, ensemble methods, and ANN are capable of handling large datasets and many features quite well. The linear and logistic regression generally perform better than other models in case the size of the dataset is small. Application of variable reduction techniques (shown in Chapter 7) enables the linear models to handle large datasets. The performance of ANN increases with an increase in the size of the dataset.
Given linear regression, logistic regression, and CART are relatively simpler models, they have better model interpretation as compared to the ensemble models and ANN.
Model Trade-off
Often, it’s a trade-off between different factors when selecting a model. ANN, SVM, and some ensemble methods can be used to create very accurate predictive models, but they may lack simplicity and interpretability and may take a significant amount of resources to train.
In terms of selecting the final model, models with lower interpretability may be preferred when predictive performance is the most important goal, and it’s not necessary to explain how the model works and makes predictions. In some cases, however, model interpretability is mandatory.
Interpretability-driven examples are often seen in the financial industry. In many cases, choosing a machine learning algorithm has less to do with the optimization or the technical aspects of the algorithm and more to do with business decisions. Suppose a machine learning algorithm is used to accept or reject an individual’s credit card application. If the applicant is rejected and decides to file a complaint or take legal action, the financial institution will need to explain how that decision was made. While that can be nearly impossible for ANN, it’s relatively straightforward for decision tree–based models.
Different classes of models are good at modeling different types of underlying patterns in data. So a good first step is to quickly test out a few different classes of models to know which ones capture the underlying structure of the dataset most efficiently. We will follow this approach while performing model selection in all our supervised learning–based case studies.
Chapter Summary
In this chapter, we discussed the importance of supervised learning models in finance, followed by a brief introduction to several supervised learning models, including linear and logistic regression, SVM, decision trees, ensemble, KNN, LDA, and ANN. We demonstrated training and tuning of these models in a few lines of code using sklearn and Keras libraries.
We discussed the most common error metrics for regression and classification models, explained the bias-variance trade-off, and illustrated the various tools for managing the model selection process using cross validation.
We introduced the strengths and weaknesses of each model and discussed the factors to consider when selecting the best model. We also discussed the trade-off between model performance and interpretability.
In the following chapter, we will dive into the case studies for regression and classification. All case studies in the next two chapters leverage the concepts presented in this chapter and in the previous two chapters.
Cross validation will be covered in detail later in this chapter.
See the activation function section of Chapter 3 for details on the sigmoid function.
MLE is a method of estimating the parameters of a probability distribution so that under the assumed statistical model the observed data is most probable.
The approach of projecting data is similar to the PCA algorithm discussed in Chapter 7.
Bias and variance are described in detail later in this chapter.
Split is the process of converting a nonhomogeneous parent node into two homogeneous child nodes best possible).
In this table we do not include AdaBoost and extra trees as their overall behavior across all the parameters are similar to Gradient Boosting and Random Forest, respectively.