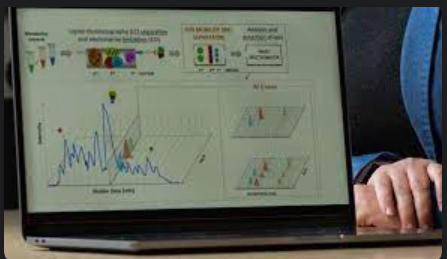
We developed and demonstrated a new metabolomics workflow for studying engineered microbes in synthetic biology applications. Our workflow combines state-of-the-art analytical instrumentation that generates information-rich data with a novel machine learning (ML)-based algorithm tailored to process it.
In our roles as Pacific Northwest National Laboratory (PNNL) scientists, we led this multi-institutional study, which was published in Nature Communications.
Addressing the challenges of complex samples
Metabolites are small molecules produced by large networks of cellular processes and biochemical reactions in living systems. The sheer diversity of metabolite classes and structures constitutes a significant analytical challenge in terms of detection and annotation in complex samples.
Analytical instrumentation able to analyze hundreds of samples in ever faster and more accurate ways is critical in various metabolomics applications, including the development of microorganisms that can produce desirable fuels and chemicals in a sustainable way.
Multidimensional measurements using liquid chromatography (LC), ion mobility and data-independent acquisition mass spectrometry (MS) improve metabolite detection by linking the separations in a single analytical platform. The potential for metabolomics has been previously demonstrated, but this kind of multidimensional information-rich data is complex and cannot be processed with traditional tools. Therefore, algorithms and software tools capable of processing it to extract accurate metabolite information are needed.
Richer data requires software upgrades to match
We optimized a combination of sophisticated instruments for fast analyses and generated multidimensional data, rich in information that can be used to tease apart complex metabolomes.
For the computational method, Dr. Bilbao created a new algorithm, called PeakDecoder, to enable interpretation of the multidimensional data and ultimately identify individual molecules in complex mixtures. Our algorithm learns to distinguish true co-elution and co-mobility directly from the raw data of the studied samples and calculates error rates for metabolite identification. To train the ML model, it proposes a novel method to generate training examples, similar to the target-decoy strategy commonly used in proteomics. Once the model is trained, it can be used to score metabolites of interest from a library with an associated false discovery rate. And contrary to existing methods, it can also be used with libraries of small size.
The key outcomes of the paper were:
- An optimized, fast analytical method for metabolites using LC, ion mobility and MS
- A new algorithm enabling the processing of multidimensional MS data and estimation of error rates in metabolomics
Large-scale and accurate metabolomics profiling
The method takes a third of the sample analysis time of previous conventional approaches by using optimized LC conditions. PeakDecoder enables accurate profiling in multidimensional MS measurements for large scale studies.
We used the workflow to study metabolites of various strains of microorganisms engineered by the Agile BioFoundry to make various bioproducts, such as polymers and diesel fuel precursors. We were able to interpret 2,683 metabolite features across 116 microbial samples.
“This metabolomics capability has far-reaching benefits, beyond synthetic biology, across environmental and biological research.” – Dr. Kristin Burnum-Johnson, biochemist and Agile BioFoundry TEST task lead.
However, it should be noted that the current algorithm is not fully automated due to software dependencies and requires a metabolite library acquired with compatible analytical conditions for inference.
Empowering PeakDecoder with AI
We are working on the next version of the algorithm leveraging advanced artificial intelligence (AI) methods used in other fields, such as computer vision. A user-friendly and fully automated version of PeakDecoder will support other types of molecular profiling workflows, including proteomics and lipidomics. Performance will be evaluated with more types of experimental data and AI-predicted multidimensional molecular libraries. The new version is expected to provide significant advances for multiomics research.
“Advanced AI-based software could potentially replace traditional MS tools that require heavy human intervention” – Dr. Aivett Bilbao, computational scientist.